Characterization of the core bacteriobiome in the rhizosphere of greenhouse vegetables: taxonomic diversity and putative functions
DOI:
https://doi.org/10.31251/pos.v3i3.128Ключевые слова:
rhizosphere microbiota, 16S rRNA gene amplicon sequencing, bitter melon, wax gourd, kiwano, cowpea, West SiberiaАннотация
The aim of the study. The aim was to profile 16S rRNA gene diversity and to assess functional potential of bacterial assemblages in the rhizosphere of some unconventional vegetables grown in protected greenhouse conditions in West Siberia.
Location and time of the study. Novosibirsk, Russia, 2016.
Methodology. At the end of the growing season in the middle of September the rhizosphere soil was collected from the plants of wax gourd (Benincasa hispida), bitter melon (Momordica charantia), kiwano (Cucumis metuliferus) and cowpea (Vigna unguiculata) grown on peat-based substrate in a polyethylene-protected greenhouse that has been in operation for more than 40 years. The metagenomic DNA was extracted and amplified with V3-V4 primers for 16S rRNA genes, and the amplicons sequenced with Illumina MiSeq. The obtained OTUs tables were used to predict putative functions by running through the FAPROTAX database.
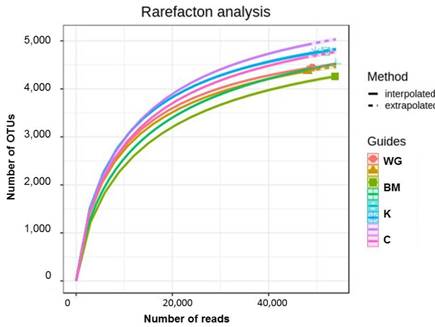
Main results. The rhizosphere bacteriobiome was dominated by Proteobacteria (32±11% of the total number of sequence reads), Acidobacteria (23±7%) and Actinobacteria (18±3%) phyla, together accounting for about three quarters of the rhizosphere bacteriobiome. In total 20 bacterial phyla were found. The rhizosphere bacteriobiome was surprisingly diverse with Shannon index ranging 7.0–7.5. The number of the observed operational taxonomic units (OTUs) per sample was very high, ranging 4,500–4,900, and the potential number of OTUs estimated as 5,100–5,700; all those OTUs were evenly and equitably represented in the bacteriobiome, and dominance indices (Simpson dominance and Berger-Parker) were very low. The main dominant OTU represented Bradyrhizobiaceae family and accounted for just 1% on average. Overall the study identified 27 OTUs belonging to the Bradyrhizobiaceae family, but only four of them were ascribed to nitrogen fixation by FAPROTAX. Function prediction by FAPROTAX also suggested that bacteriobiome had a marked potential for the carbon cycle, denitrification, aromatic compound and plant polymer degradation, but no plant pathogens. The biggest difference in rhizosphere bacteriobiome diversity was observed between the bitter melon and the other three vegetable crops: bitter melon had much increased abundance of Arthrobacter and Sphingomonas as compared with wax gourd, kiwano and cowpea, and increased number of bacterial species associated with aromatic compounds degradation.
Conclusion. Based on the finding that the studied rhizosphere bacteriobiomes were very diverse, we conclude that the crops were able to recruit diverse microbiota from the peat-based soil substrate, which, in its turn, means that diverse soil substrate microbiota has been sustained over several decades of the greenhouse operation. All crops apparently shaped distinct bacteriobiomes in their rhizosphere, which ideally should be included into studies of plant-associated bacterial diversity profiles for breeding and sustainable production.
Скачивания
Загрузки
Опубликован
Как цитировать
Выпуск
Раздел
Лицензия
Copyright (c) 2021 Почвы и окружающая среда

Это произведение доступно по лицензии Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.



