
CHARACTERIZATION OF THE CORE BACTERIOBIOME IN THE RHIZOSPHERE OF GREENHOUSE VEGETABLES: TAXONOMIC DIVERSITY AND PUTATIVE FUNCTIONS
N. B. Naumova 1
 , O. A. Savenkov1 , T. Yu. Alikina2 , Yu.V.Fotev3
, O. A. Savenkov1 , T. Yu. Alikina2 , Yu.V.Fotev3
Address: 1Institute of Soil Science and Agrochemistry SB RAS, Lavrentieva, 8/2, Novosibirsk, 630090, Russia
2Institute of Chemical Biology and Fundamental Medicine SB RAS, Lavrentieva 8, Novosibirsk 630090, Russia
3Central Siberian Botanical Garden, SB RAS, Zolotodolinskaya 101, 630090, Novosibirsk, Russia
The aim of the study. The aim was to profile 16S rRNA gene diversity and to assess functional potential of bacterial assemblages in the rhizosphere of some unconventional vegetables grown in protected greenhouse conditions in West Siberia.
Location and time of the study. Novosibirsk, Russia, 2016.
Methodology. At the end of the growing season in the middle of September the rhizosphere soil was collected from the plants of wax gourd (Benincasa hispida), bitter melon (Momordica charantia), kiwano (Cucumis metuliferus) and cowpea (Vigna unguiculata) grown on peat-based substrate in a polyethylene-protected greenhouse that has been in operation for more than 40 years. The metagenomic DNA was extracted and amplified with V3-V4 primers for 16S rRNA genes, and the amplicons sequenced with Illumina MiSeq. The obtained OTUs tables were used to predict putative functions by running through the FAPROTAX database.
Main results. The rhizosphere bacteriobiome was dominated by Proteobacteria (32±11% of the total number of sequence reads), Acidobacteria (23±7%) and Actinobacteria (18±3%) phyla, together accounting for about three quarters of the rhizosphere bacteriobiome. In total 20 bacterial phyla were found. The rhizosphere bacteriobiome was surprisingly diverse with Shannon index ranging 7.0–7.5. The number of the observed operational taxonomic units (OTUs) per sample was very high, ranging 4,500–4,900, and the potential number of OTUs estimated as 5,100–5,700; all those OTUs were evenly and equitably represented in the bacteriobiome, and dominance indices (Simpson dominance and Berger-Parker) were very low. The main dominant OTU represented Bradyrhizobiaceae family and accounted for just 1% on average. Overall the study identified 27 OTUs belonging to the Bradyrhizobiaceae family, but only four of them were ascribed to nitrogen fixation by FAPROTAX. Function prediction by FAPROTAX also suggested that bacteriobiome had a marked potential for the carbon cycle, denitrification, aromatic compound and plant polymer degradation, but no plant pathogens. The biggest difference in rhizosphere bacteriobiome diversity was observed between the bitter melon and the other three vegetable crops: bitter melon had much increased abundance of Arthrobacter and Sphingomonas as compared with wax gourd, kiwano and cowpea, and increased number of bacterial species associated with aromatic compounds degradation.
Conclusion. Based on the finding that the studied rhizosphere bacteriobiomes were very diverse, we conclude that the crops were able to recruit diverse microbiota from the peat-based soil substrate, which, in its turn, means that diverse soil substrate microbiota has been sustained over several decades of the greenhouse operation. All crops apparently shaped distinct bacteriobiomes in their rhizosphere, which ideally should be included into studies of plant-associated bacterial diversity profiles for breeding and sustainable production.
Keywords:
rhizosphere microbiota; 16S rRNA gene amplicon sequencing; bitter melon; wax gourd; kiwano; cowpea; West SiberiaHow to cite: Naumova N. B., Savenkov O.A., Alikina T.Yu., Fotev Yu.V. Characterization of the core bacteriobiome in the rhizosphere of greenhouse vegetables: taxonomic diversity and putative functions // The Journal of Soils and Environment. 2020. 3(3). e128. 10.31251/pos.v3i3.128
Introduction
Sustainable crop production is of utmost importance for providing food, bioenergy, timber and other plant materials for the ever-growing human population. Except for carbon, plants derive macro- and micronutrients from soil, affecting the latter by their rhizodeposition and aboveground litter. Rhizosphere is the primary interface between plant and soil. The rhizosphere microbiota has been recognized as an important factor regulating the homeostasis of the host plant and influencing its fitness, and, ultimately, determining the quantity and quality of phytomass production. Thus understanding plant-microbe interactions is indispensable for maintaining, increasing or restoring plant health (Berendsen et al. 2012; Trivedi et al., 2017) and agricultural ecosystem productivity (de la Fuente Canto et al., 2020; Tian et al., 2020).
It is even more essential to understand the processes going on in the rhizosphere and their specifics for better utilization of the microbial diversity (Vishwakarma et al., 2020) in sustainable greenhouse production, where biotic and abiotic factors can be controlled. Further climate warming that is forecast for the Asian part of northern Eurasia, alongside the quest for functional food, invites cultivation of new unconventional crops in the region, both under protected and open field conditions (Naumova et al. 2019). Vegetables are one of the most essential components of human diets due to their high nutritional value which provides carbohydrates, proteins, vitamins, and several other useful food elements. Due to these, vegetable consumption has increased considerably. Recently such vegetable crops as wax gourd, bitter melon, horned cucumber (kiwano), cowpea and some others have increasingly attracted attention as supplementary sources of healthy and functional food (Fang et al., 2019; Huang et al., 2020; Zhu et al., 2021), also in the Novosibirsk region (Fotev et al., 2019). Under protected condition the crops grow on artificial substrates, commonly based on peat. However, except for some staple vegetable crops like potato, tomato and cucumber, there is few data about the core and total rhizosphere bacteriobiome of many vegetable crops and about specific mechanisms of how plants shape it (Kumar, Dubei, 2020). Such knowledge is particularly important for crops grown in protected conditions in greenhouses, especially operated for long time, as ultimately it will contribute to better crop performance and higher produce quality. This actualizes studies of unconventional vegetable crops’ growth and development in the protected field in the Novosibirsk region. The aim of this pilot study was to profile 16S rRNA gene diversity and to assess functional potential of bacterial assemblages in the rhizosphere of the some unconventional vegetables grown in protected greenhouse conditions in West Siberia.
Experimental setup
The greenhouse (54°48'46.7"N, 83°06'10.2" E, Figure S1) was constructed in 1976 by removing the top 50 cm layer of natural Phaeozem, filling the void by 10 cm of sand and a mixture of peat and sawdust to level with the surface. Since then the greenhouse has been used for breeding vegetable crops and maintaining their seed collections, as well as for introducing new for Russia vegetables. Over the years once in a while (2-3 years) the same or similar substrate was added as needed and mineral fertilizers (NPK) applied at crop-dependent rate during the growing season. In 2016, i.e. the year of sampling for the study, the soil substrate had neutral pH (7.0), 8.9% of organic carbon, 1.3% of organic nitrogen (C:N ratio 8.8), 154 mg N-NO3/ g soil, 77 P-P2O5 mg/g soil and 204 K-K2O mg/g soil. The polyethylene-covered greenhouse is not heated. Thus the growing season usually lasts since May till the end of September. The plants of the accessions from the Bioresource Scientific Collection of the Central Siberian Botanical Garden (CSBG SB RAS) UNU No. USU 440534 were used in the study. In the 2016 growing season the following vegetable crops were planted: wax gourd Benincasa hispida (Thunb.) Cogn., bitter melon Momordica charantia L., kiwano (horned cucumber) Cucumis metuliferus E.Mey and cowpea Vigna unguiculata (L.) Walp. at the density of 2 plants per m2; each crop on a subplot of ca. 5.3 m2.
Soil sampling
At the very end of the growing season (Figure S2, A-D) four plants were chosen at random from each crop subplot; then from each two of them rhizosphere soil was collected as the soil strongly adhering to 1–3 mm thick roots after gentle shaking over polyethylene (Zhao et al. 2010) and bulked together to produce two individual samples for each crop. Thus eight individual soil samples were collected. The samples were brought into the laboratory and stored at –20°C prior to DNA extraction.
Extraction of total nucleic acid from soil
Total DNA was extracted from the soil samples using the DNA isolation Kit (DNeasy PowerSoil Kit, Qiagen, Germany) as per manufacturer’s instructions. The bead-beating was performed using TissueLyser II (Qiagen, Germany) 10 min at 30 Hz. The quality of the extracted DNA was assessed by the spectrophotomer NanoDrop ND-1000 (Thermo Fisher, USA), by agarose gel electrophoresis and pilot PCR. No further purification of the DNA was needed.
16S rRNA gene amplification and sequencing
The 16S DNA region was amplified with the primer pair V3/V4 combined with Illumina adapter sequences (Fadrosh et al. 2014). Amplification was performed by polymerase chain reaction (PCR) as described by Igolkina et al. (2018) in three replicates for one DNA sample used as template. A total of 200 ng PCR product from each PCR replicate were pooled together and purified through MinElute Gel Extraction Kit (Qiagen, Germany). The obtained libraries were sequenced with 2 × 300 bp paired-ends reagents on MiSeq (Illumina, USA) in SB RAS Genomics Core Facility (ICBFM SB RAS, Novosibirsk, Russia). The read data reported in this study were submitted to the GenBank under the study accession PRJNAPRJNA702204 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA702204).
Bioinformatics and statistical analysis
Raw sequences were analyzed with the UPARSE pipeline (Edgar, 2013) using Usearch v11.0. The UPARSE pipeline included the merging of paired reads; read quality filtering; length trimming; merging of identical reads (dereplication); discarding singleton reads; removing chimeras and operational taxonomic unit (OTU) clustering using the UNOISE-OTU algorithm (Edgar, 2016a). The OTU sequences were assigned taxonomically using the SINTAX (Edgar, 2016b) and 16S RDP training set v.16 (Wang et al., 2007). After quality filtering and chimera removal overall 8743 OTUs were found; 23 of them represented Archaea and were removed from further analyses. Taxonomic structure of thus obtained bacteriobiome, i.e. bacterial part of the microbiome which is defined by J.R. Marchesi and J. Ravel (2015) as referring “to the entire habitat, including the microorganisms (bacteria, archaea, lower and higher eurkaryotes, and viruses), their genomes (i.e., genes), and the surrounding environmental conditions”, was estimated by the ratio of the number of taxon-specific sequences reads to the total number of sequence reads (relative abundance of taxa, expressed as a percentage). The obtained OTUs tables were used to predict putative functions by running through the Functional Annotation of Prokaryotic Taxa (FAPROTAX, v.1.2.4) database (Louca et al., 2016), available at http://www.zoology.ubc.ca/louca/FAPROTAX.). Thus some of the OTUs identified in the study were assigned functionality or environment niche. Biodiversity indices calculated with the help of the PAST 3.19 software (Hammer et al., 2001). The data were represented as a mean.
Taxonomic richness of the rhizosphere bacteriobiome
After quality filtering and chimera removal overall 8720 bacterial OTUs were found in the rhizosphere samples. The 16S amplicon sequence reads datasets were analyzed by individual rarefaction (Figure 1): the number of OTUs detected, reaching a plateau with increasing number of sequences, showed that the sampling effort was close to saturation for all samples, thus being enough to compare diversity (Hughes, Hellmann 2005).
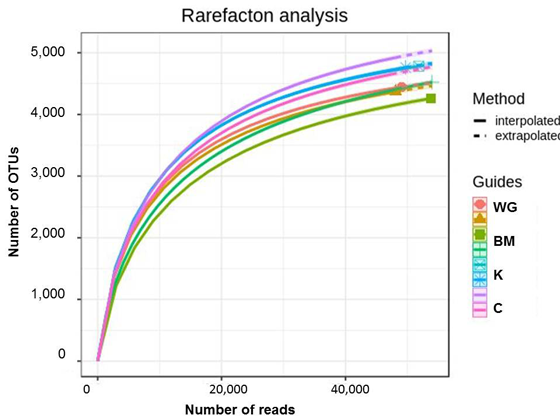
Figure 1. Rarefaction curve for the studied rhizosphere soils. The samples are not specified as all lines are close to each other.
The number of OTUs per sample averaged 4,713, ranging 4,400-5,000. In total 20 bacterial phyla and 53 identified and 15 non-identified (below the phylum level) classes were found. Quite a lot of OTU-level clusters (19% of the total number of OTUs, or 14% of the total number of sequence reads) could not be classified below the domain level, most likely signifying their absence in the database. The most OTU-rich phylum was Proteobacteria with 2,552 OTUs (29% of the total number of OTUs), followed by Acidobacteria with 1,724 OTUs (20%) and Actinobacteria, represented by 1,384 OTUs (16%).
Taxonomic structure of the rhizosphere bacteriobiome
As for the relative abundance of taxa, assessed by the ratio of a taxon-specific sequence to the total number of sequence reads, the rhizosphere bacteriobiome of the studied vegetables was dominated by Proteobacteria (32±11% of the total number of sequence reads), Acidobacteria (23±7%) and Actinobacteria (18±3%) phyla (Figure 2, A). Summed up, the joint abundance of these dominant phyla comprised more than three quarters of the rhizosphere bacteriobiome. Other prevailing phylum was Bacteroidetes with its 4.3±1.6%. The phyla Gemmatimonadetes, Firmicutes and cand.Saccharibacteria each accounted for ca. 1%, being minor dominants. The rest of the identified phyla were quite negligible, contributing less than 1.0% each. Unclassified Bacteria, however, comprised 16% on average.
At the class level Actinobacteria, averaging 17.6±3.2% was the most abundant (Figure 2, B), followed by Alphaproteobacteria (15.4±5.3%) and Acidobacteria group 6 (14.2±5.2%). Betaproteobacteria and Deltaproteobacteria contributed slightly over 2% each into the total bacteriobiome. Other Acidobacteria classes, i.e. groups 16, 3 and 4, accounted for 1-4% each. At the order level, some unclassified Bacteria were the most abundant, followed by Acidobacteria group 6 Rhizobiales, Actinomycetales, Gaiellales and unclassified Actinobactria (Figure 2, C).
At the OTUs level, the structure was very even and equitable: only 7 OTUs had relative abundance ranging from 0.5 to 1.1%. The main dominant OTU represented Bradyrhizobiaceae family and accounted for just 1.1%. Other dominants represented Spartobacteria class (one OTU with 0.8%), Acidobacteria group 16 (one OTU with 0.7%), Sphagnomonas genus of the Alphaproteobacteria (two OTUs with 0.5‒0.7%) and Actinobacteriales and Gaiellales orders of Actinobacteria class (also two OTUs with 0.5‒0.7%). Overall such low relative abundance for an OTU together with a very high number of OTUs detected resulted in very high diversity indices and extremely low dominance indices (Table 1).
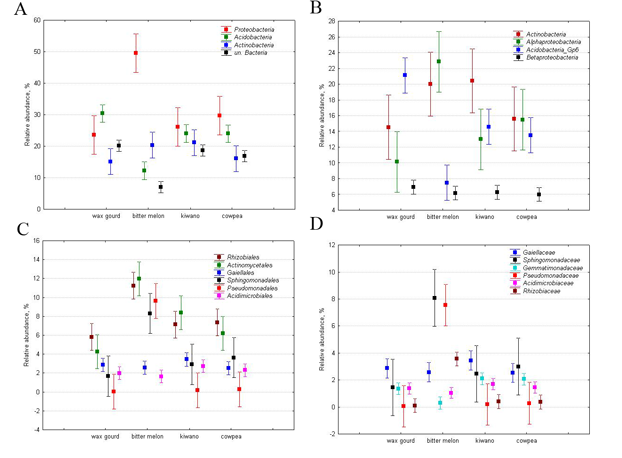
Figure 2. Relative abundance of the major dominant bacterial taxa: A – phyla, B – classes, C – orders and D – families. The taxa that were not explicitly assigned a taxonomy at the corresponding level, are not shown. The markers show the means, and the whiskers show standard deviation.
Table 1. Bacteriobiome α-diversity indices in the rhizosphere soil of vegetables grown in a greenhouse
| Vegetable crop | Observed OTUs richness | Chao-1 | Shannon | Dominance | Berger-Parker | Evenness |
|---|---|---|---|---|---|---|
| Wax gourd | 4515±62 a* | 5162±33 a | 7.52±0.03 b | 0.001±0.000 a | 0.008±0.000 a | 0.41±0.01 b |
| Bitter melon | 4526±167 a | 5353±213 ab | 6.96±0.13 a | 0.005±0.000 b | 0.036±0.004 c | 0.23±0.02 a |
| Kiwano | 4900±18 b | 5551±49 bc | 7.62±0.02 b | 0.001±0.000 a | 0.013±0.003 ab | 0.42±0.01 b |
| Cowpea | 4911±200 b | 5730±187 c | 7.53±0.06 b | 0.001±0.000 a | 0.016±0.001 b | 0.38±0.01 b |
*Different letters in columns indicate that the values differ at P≤0.05 level (LSD test).
Principal components analysis revealed some separation of soil samples from different crops, the replicates being close to each other. Similar pattern of sample relationship was obtained by extracting principal components from one data matrix with OTUs’ relative abundance as variables for analysis and from another data matrix with the number of OTUs assigned to a certain function/niche in the FAPROTAX database as variables for analysis. In the first case (Figure 1, A), most of the variance in PC 1 was due to the Arthrobacter sp.(Actinobacteria) and OTUs, belonging to Acidobacteria Group_16 and Sphingomonas genus, at the negative pole (together with bitter melon), and unclassified OTUs, belonging to Actinomycetales, Acidobacteria Group_6 and Bacteria, at the PC 1 positive pole (together with wax gourd, kiwano and cowpea).
Putative functions in the rhizosphere bacteriobiome
At the date of analysis the FAPROTAX database contained information about 92 functional groups. The overwhelming majority of the total number of OTUs, precisely 7574, or. ca. 87%) found in this study, could not be assigned to any of those groups. The rest 1146 OTUs were assigned to 50 groups, with some of them being assigned to more than one functional group. Overall function prediction by FAPROTAX suggested that the core bacteriobiome had a marked potential for the carbon cycle, nitrate metabolism, aromatic compound degradation, cellulolysis, and xylanolysis, but no plant pathogens and few human ones. The principal components analysis of the data matrix with the number of OTUs (not their relative abundance), assigned to specific functions, as columns, i.e. variables for the analysis, and rhizosphere samples as rows, i.e. cases, revealed (Figure 1, B) that most of the data variance in PC 1 was due to the chemoheterotrophy, aromatic compounds degradation and cellulose and chitin lysis at the negative pole (together with bitter melon samples), and mineral N, C and S transformation and xylanolysis at the PC 1 positive pole (together with wax gourd, kiwano and cowpea). Naturally, chemoheterotrophy was by far the most OTU rich function. As nitrogen is one of the most important plant nutrients in peat-based soil substrates, so the number of OTUs ascribed to nitrogen-related functions was of special interest. Nitrate reduction was second-ranked in species richness, with 91 OTUs of Gaiella occulta (Actonobacteria phylum), 32 OTUs of the Proteobacteria phylum, 14 OTUs of Opitutus terrae of the Verrucomicrobia phylum. Nitrification potential was ascribed to 35 OTUs: 25 of unidentified Nitrospira (Nitrospirae phylum) and 9 of Nitrosospira (Proteobacteria phylum). Nitrogen fixation was identified for six OTUs of the Proteobacteria phylum, 4 of them representing Bradyrhizobium genus, one Beijerinckia sp., and another one Azotobacter salinestris.
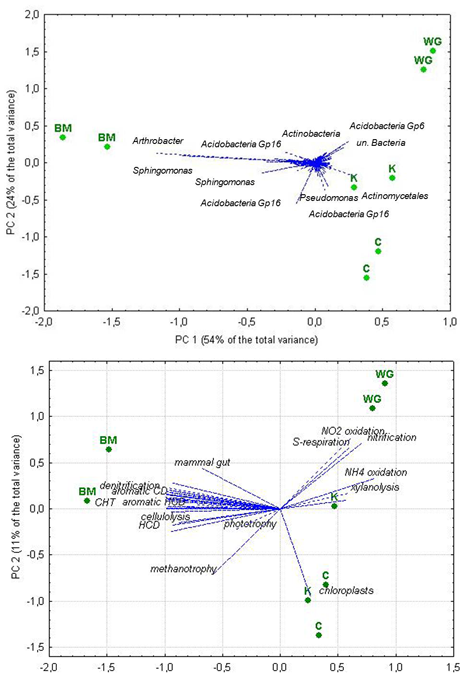
Figure 3. Principal components analysis of the data matrices with relative abundance of bacterial taxa (A) and with the number of OTUs assigned to specific functions (B) as variables for analysis: location of variables and rhizosphere soil samples in the plane of the first two principal components. Abbreviations used: WG – wax gourd, BM – bitter melon, K – kiwano, C – cowpea; CHT – chemoheterotropy; HCD – hydrocarbon degradation.
Discussion
By examining bacterial 16S rRNA gene sequence diversity we found that the rhizosphere bacteriobiome of the studied vegetables was dominated by Proteobacteria, Acidobacteria and Actinobacteria, together accounting for ca. 75% of the total number of sequence reads. We could not find information about bacteriobiome diversity in the rhizosphere soil of the same vegetable crops or in the bulk soil where they grow. Although one can find some articles allegedly devoted to bacterial diversity in the rhizosphere of vegetable crops, they described culturable bacteria, often without taxonomic attribution (Singh et al., 2017). Therefore we can compare the rhizosphere bacterial profiles obtained in our study with similar studies of more economically important and hence better studied vegetables crops like cucumber. The latter was reported to have similar composition of rhizosphere bacteriobiome at the phylum-level (the three of them together accounting for ca.70%), but Actinobacteria was second-ranked in relative abundance (Wang et al., 2018). Yet another study from China about the rhizosphere bacterial diversity of cucumber plants grown in soils covering a wide range of cucumber cropping histories and environmental conditions found that the three main dominant bacterial phyla were Proteobacteria, Bacteroidetes and Actinobacteria (Tian, Gao, 2014). Comparison with the rhizosphere bacteriobiome of husk tomato, grown in the same year in the same region (Naumova et al., 2019), but on loamy arable Phaeozem in the open field, showed that there the bacteriobiome structure, at least at the higher taxonomic levels, was quite similar to the found in this study. The same phylum-level bacteriobiome structure was also found in the rhizosphere of sugarcane (Zhao et al., 2020) and canola (Floc'h et al., 2020).
Notably, FAPROTAX found nitrate transformation functions, namely denitrification/nitrification, to be the most species-rich (after chemoheterotrohy), which is quite reasonable in the greenhouse soil substrate receiving mineral NPK fertilizers with nitrogen as nitrate. We did not find plant pathogenic bacteria in the entire plethora of OTUs identified in the rhizosphere of vegetables. Together with the fact that no disease afflicted the plants during the growing season at the end of which the sampling was performed, it suggests that the greenhouse soil substrate is either disease-suppressive (Berendsen et al., 2012; Trivedi et al., 2017), or just lacks the pathogens for these rather novel crops. However, partially this finding resulted from the incompleteness of FAPROTAX, as there are many yet unidentified and unculturable bacteria. It should be emphasized that the database has been initiated “by classifying >30,000 marine microorganisms into metabolic functional groups” (Louca et al., 2016, p. 1272), which may explain that a) no plant pathogens were found, and b) very low percentage (13%) of the total number of OTUs assigned to some functions/niches. Such low percentage may imply that the database does not represent adequately soil bacteria. Moreover, there is a basic methodological flaw in the database, as there is no consistency in the terminology used by the authors to describe metabolic functions: for instance, “chemoheterotrophy” alongside with “plant pathogens”, “aromatic_compound_degradation” alongside with “human gut”, etc. From these two examples it is obvious that the predicted functions are not exclusive. In addition, functional groups there are often nested: for example, all taxa associated with aerobic chemoheterotrophy are also within chemoheterotrophy. It raises another issue about how relative abundance of OTUs assigned to certain functions can be calculated, which is what many researchers did (as an example see Zhou et al., 2020). Because of this we analyzed only the number of OTUs for certain functions, rather than their relative abundance, to get a glimpse of the database performance with soil data: we tend to conclude that currently the FAPROTAX output is quite limited and often confusing to interpret. Moreover, one should be well aware that the number of OTUs, allotted to certain functions/environmental niches by the FAPROTAX database, is indicative only of a certain potential functionality and by no means of the actual respective functional performance and/or functional bacterial assemblage structure.
Although the definition of rhizosphere as the volume of soil affected by exudates from plant root tissues (Pinton et al., 2007) is rather straightforward and unequivocal, operationally it is rather challenging to determine the boundaries of thus affected soil volume and collect soil samples appropriately. Besides, the rhizodeposition may differ between roots of different age/thickness, the effect varying between different cultivars/crops/species (Rovira, 1959; Singh et al., 2017); all these factors may add to the variation among rhizosphere microbiomes. With this in mind, we believe that close location of the two individual rhizosphere soil samples from each crop in the plane of the first two principal components (Figure 2) may indicate good replication and low intra-crop variability. Notably, among the studied samples we found the similar pattern of relationship, obtained by principal components analysis of the two data matrices with different kind of variables: 1) the matrix with relative abundance of OTU’s sequences in the total number of sequences, generated in the study, and 2) the number of OTUs ascribed to a certain function/niche in the FAPROTAX database. As those variables are by no means correlated, the similarity in the samples’ relationship pattern confirms differences in functional and taxonomic diversity between bitter melon and other crops in this study.
Our finding that bacteriobiome variation within one family (Cucurbitaceae) may be much more pronounced than the variation between different families (Cucurbitaceae and Fabaceae) implies that variation in crops’ phytomass chemistry, and in particular between bitter melon and wax gourd (Yu et al., 2019; Zhu et al., 2021), may be the factor, primarily responsible for shaping the bacteriobiome diversity and consequently, the revealed pattern of the studied samples relationship. The spectra of cucurbitacins, a group of bitter and highly oxygenated tetracyclic triterpenes that are produced by cucurbits, although structurally related, but yet unique species-specific products (Zhou et al., 2016); they might contribute to shaping not only the entire bacteriobiome, but also to the absence of plant pathogenic species.
We found a surprisingly diverse bacteriobiome in the rhizosphere of some vegetable crops grown on peat substrate in polyethylene-protected greenhouse that has been in operation for more than 40 years. The α-diversity indices, calculated in this study, are comparable with the indices calculated for the husk tomato grown in the open field in the same region (Naumova et al., 2019); for the rhizosphere bacteriobiome of other vegetables, like cucumber (Wen et al., 2020) and other crops (sugarcane, Zhao et al., 2020). Therefore it seems that the chemical diversity of root exudates (Rovira, 1969; Uren, 2000; Zhalnina et al., 2018), i.e. compounds, easily available for microbial utilization, can recruit and sustain high bacterial species diversity in rhizosphere even after the long-term use for greenhouse crop production. As reduction in soil microbial diversity was shown to be associated with the burst of soil-borne plant diseases (Mazzola et al., 2004), the high biodiversity in this study apparently contributed to healthy status of the vegetables.
One should always bear in mind that proportions of gene copy numbers, be they 16S rRNA or functional genes, are not entirely synonymic with the number of the relevant organisms present and the intensity of the processes they perform. In fact, few publications report revealing microbial community structure-process links (Bier et al., 2015). Therefore microbiome profiles provide just scaffolding for constructing more comprehensive and/or targeted research.
Conclusion
To the best of our knowledge, here we present the first study describing bacteriobiome in the rhizosphere of several unconventional for West Siberia vegetable crops, such as bitter melon, wax gourd, kiwano and cowpea, grown on peat-based soil substrate in the greenhouse, using high-throughput sequencing techniques. We believe the obtained information will help focusing further more extensive research on rhizosphere microbiome as related to plant host and soil environment and also serve as a reference for comparing other data, as well as for meta-analysis. Based on the finding that the studied rhizosphere bacteriobiomes were very diverse, we conclude that the crops were able to recruit diverse microbiota from the peat-based soil substrate, which, in its turn, means that diverse microbiota has been sustained over several decades of the greenhouse operation. All crops apparently shaped distinct bacteriobiomes in their rhizosphere, which ideally should be included into studies of plant-associated bacterial diversity profiles for breeding and sustainable greenhouse production. As an important plant-associated characteristic, rhizosphere microbiota deserves more comprehensive research by combined metagenomic and general microbiological techniques, which will most likely find novel taxa and/or novel ecotypes and relate them to certain functions. We believe that such knowledge will be extremely helpful in the future for shaping plant-associated artificial microbiota for quality yields of functional food production under hydroponics conditions.
Financial support
The study was financially supported by the Ministry of Science and Higher Education of the Russian Federation (projects nos. АААА-А17-117030110078-1, АААА-А17-117012610054-6 and АААА-А17-117020210021-7).
References
Berendsen R.L., Pieterse C.M., Bakker P.A. The rhizosphere microbiome and plant health, Trends Plant Sci., 2012, V.17, No.8, p. 478–486.
Bier R.L., Bernhardt E.S., Boot C.M., Graham E.B., Hall E.K., Lennon J.T., Nemergut D.R., Osborne B.B., Ruiz-González C., Schimel J.P., Waldrop M.P., Wallenstein M. D. Linking microbial community structure and microbial processes: an empirical and conceptual overview, FEMS Microbiol. Ecol., 2015, V.91, No. 10, fiv113.
Edgar R.C. UPARSE: highly accurate OTU sequences from microbial amplicon reads, Nat Methods, 2013, V.10, p. 996–998.
Edgar R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon reads, bioRxiv, 2016a.
Edgar R.C. SINTAX, a Simple Non-Bayesian Taxonomy Classifier for 16S and ITS Sequences, bioRxiv, 2016b.
Fadrosh D.W., Ma B., Gajer P., et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform, Microbiome, 2014, V.2, P.1–6.
Fang E.F., Froetscher L., Scheibye-Knudsen M., Bohr V.A., Wong J.H., Ng T.B. Emerging Antitumor Activities of the Bitter Melon (Momordica charantia), Curr. Protein Peptide Sci., 2019, V.20, No. 3, p.296–301.
Floc'h J.B., Hamel C., Lupwayi N., Harker K.N., Hijri M., St-Arnaud M. Bacterial Communities of the Canola Rhizosphere: Network Analysis Reveals a Core Bacterium Shaping Microbial Interactions, Front Microbiol., 2020, V. 11, Article 1587.
Fotev Y.V., Syso A.I., Shevchuk O.M. Introduction in Siberia (Russia) of new vegetable species with a high biochemical value, In book: PlantGen2019. Current. Challenges in Plant Genetics, Genomics, Bioinformatics, and Biotechnology. Proc. of the Fifth Inter. Sci. Conf. (Novosibirsk, 24-29 June, 2019). Kochetov A., Salina E. (eds.). Novosibirsk, 2019. 12–14.
de la Fuente Cantó C., Simonin M., King E., Moulin L., Bennett M.J., Castrillo G., Laplaze L. An extended root phenotype: the rhizosphere, its formation and impacts on plant fitness, The Plant J., 2020, V. 103, Iss. 3, p. 951–964.
Hammer O., Harper D.A.T., Ryan P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis, Palaeontologia Electronica, 2001, V.4, Iss.1, p.9.
Huang H., Chen F., Long R., Huang G. The antioxidant activities in vivo of bitter gourd polysaccharide, // Int. J. Biol. Macromol., 2020, V.145, p.141–144.
Igolkina А.А., Grekhov G.A., Pershina E.V., et al. Identifying components of mixed and contaminated soil samples by detecting specific signatures of control 16S rRNA libraries, Ecol. Ind., 2018, V.94, p. 446–453.
Kumar A., Dubei A. Rhizosphere microbiome: Engineering bacterial competitiveness for enhancing crop production, J. Adv. Res., 2020, V. 24, p.337–352.
Louca S., Parfrey L.W., Doebeli M. Decoupling function and taxonomy in the global ocean microbiome, Science, 2016, V. 353, p.1272–1277.
Marchesi J.R., Ravel J. The vocabulary of microbiome research: a proposal, Microbiome, 2015, V. 3, p. 31.
Mazzola M. Assessment and management of soil microbial community structure for disease suppression, Annu Rev Phytopathol., 2004, V.42, P.35–59.
Pinton R., Varanini Z., Nannipieri P. The Rhizosphere: Biochemistry and Organic Substances at the Soil-Plant Interface: Boca Raton: CRC Press, 2007.
Rovira A.D. Plant root exudates, Botanical Review, 1959, V.35, No. 1, p. 35–57.
Rovira A.D. Plant root excretions in relation to the rhizosphere effect. IV. Influence of plant species, age of plant, light, temperature, and calcium nutrition on exudation, Plant and Soil, 1969, V.11, p. 53–64.
Singh R., Kumar A., Singh M., Vandana, Kumar Maurya S., Pandey K.D. Microbial Diversity in the Rhizosphere of Momordica charantia L. Bitter Gourd, Int. J. Curr. Microbiol. App. Sci., 2017, V.6, No. 2, p. 67–76.
Sur S., Ray R.B. Bitter Melon (Momordica Charantia), a Nutraceutical Approach for Cancer Prevention and Therapy, Cancers, 2020, V.12, No. 8, Article 2064.
Tian Y., Gao L. Bacterial Diversity in the Rhizosphere of Cucumbers Grown in Soils Covering a Wide Range of Cucumber Cropping Histories and Environmental Conditions, Microb. Ecol., 2014, V.68, p. 794–806.
Tian L., Lin X., Tian J., Ji L., Chen Y., Tran L. P., Tian C. Research Advances of Beneficial Microbiota Associated with Crop Plants, Int. J. Mol. Sci., 2020, V.21, No. 5, p.1792.
Trivedi P., Delgado-Baquerizo M., Trivedi C., Hamonts K., Anderson I.C., Singh B.K. Keystone microbial taxa regulate the invasion of a fungal pathogen in agro-ecosystems, Soil Biol. Biochem., 2017, V.111, p.10–14.
Trivedi P., Schenk P. M., Wallenstein M. D., Singh B. K. Tiny Microbes, Big Yields: enhancing food crop production with biological solutions, Microb. Biotechnol., 2017, V.10, No. 5, p.999–1003.
Uren N.C. Types, amounts, and possible functions of compounds released into the rhizosphere by soil-grown plants. In The Rhizosphere. CRC Press: Boca Raton, USA, 2000, p. 35–56.
Vishwakarma K., Kumar N., Shandilya C., Mohapatra S., Bhayana S., Varma A. Revisiting Plant–Microbe Interactions and Microbial Consortia Application for Enhancing Sustainable Agriculture: A Review, Front. Microbiol., 2020, V. 11, Article 560406.
Wang Z., Zhang J., Wu F., Zhou X. Changes in rhizosphere microbial communities in potted cucumber seedlings treated with syringic acid, PLoS ONE, 2018, V. 13(6), e0200007.
Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naḯve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy, Appl. Environ. Microbiol., 2007, V.73, p.5261–5267.
Wen T., Yuan J., He X. et al. Enrichment of beneficial cucumber rhizosphere microbes mediated by organic acid secretion, Hortic. Res., 2020, V.7, p.154.
Yu D., Ma Y., Chen M., Dong X. KOH activation of wax gourd-derived carbon materials with high porosity and heteroatom content for aqueous or all-solid-state, J. Colloid Interface Sci., 2019, V.537, p.569–578.
Zhalnina K., Louie K.B., Hao Z., Mansoori N., da Rocha U.N., Shi S., Cho H., Karaoz U., Loqué D., Bowen B.P., Firestone M.K., Northen T.R., Brodie E.L. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly, Nat. Microbiol., 2018, V.3, No.4, p.470-480.
Zhao X., Jiang Y., Liu Q., Yang H., Wang Z., Zhang M. Effects of Drought-Tolerant Ea-DREB2B Transgenic Sugarcane on Bacterial Communities in Soil, Front Microbiol., 2020, V.11, Article 704.
Zhou Y., Ma Y., Zeng J., Duan L., Xue X., Wang H., Lin T., Liu Z., Zeng K., Zhong Y., Zhang S., Hu Q., Liu M., Zhang H., Reed J., Moses T., Liu X., Huang P., Qing Z., Liu X., … Huang, S. Convergence and divergence of bitterness biosynthesis and regulation in Cucurbitaceae, Nat. Plants., 2016, V.2, 16183.
Zhou J., Yu L., Zhang J., Zhang X., Xue Y., Liu J., Zou X. Characterization of the core microbiome in tobacco leaves during aging, Microbiology Open, 2020, V.9, No.3, e984.
Zhu M., Huang R., Wen P., Song Y., He B., Tan J., Hao H., Wang H. Structural characterization and immunological activity of pectin polysaccharide from kiwano (Cucumis metuliferus) peels, Carbohydrate polymers, 2021, V.254, Art. 117371.
Received 27 January 2021; accepted 17 February 2021; published 19 February
About the authors:
Naumova Natalia B. – Candidate of Biological Sciences, Leading Researcher in the Laboratory of Agrochemistry of the Institute of Soil Science and Agrochemistry of the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia; naumova@issa-siberia.ru
Savenkov Oleg A. – Candidate of Biological Sciences, Researcher in the Laboratory of Agrochemistry of the Institute of Soil Science and Agrochemistry of the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia; savenkov@issa-siberia.ru
Аlikina Tatiana Yu. – Junior Researcher in the “Genomics” Core Facility, Institute of Chemical Biology and Fundamental Medicine of the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia; alikina@niboch.nsc.ru
Fotev Yury V. – Candidate of Agricultural Sciences, Senior Researcher in the Laboratory of Introduction of Food Plants in the Central Siberian Botanical Garden of the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia; fotev_2009@mail.ru
The authors read and approved the final manuscript
 The article is available under Creative Commons Attribution 4.0 License
The article is available under Creative Commons Attribution 4.0 License
ХАРАКТЕРИСТИКА БАКТЕРИОБИОМА РИЗОСФЕРЫ ТЕПЛИЧНЫХ ОВОЩЕЙ: ТАКСОНОМИЧЕСКОЕ РАЗНООБРАЗИЕ И ФУНКЦИОНАЛЬНЫЕ ГРУППЫ
Н. Б. Наумова 1 , О. А. Савенков1 , Т. Ю. Аликина2 , Ю. В. Фотев3
Адрес: 1ФГБУН Институт почвоведения и агрохимии СО РАН, проспект Академика Лаврентьева, 8/2, г. Новосибирск, 630090, Россия.
2ФГБУН Институт химической биологии и фундаментальной медицины СО РАН, проспект Академика Лаврентьева, 8, г. Новосибирск, 630090, Россия
3ФГБУН Центральный Сибирский ботанический сад СО РАН, Золотодолинская, 101, г. Новосибирск, 630090, Россия
Цель исследования. Целью исследования было изучение разнообразия последовательностей генов 16S рРНК и оценка функционального потенциала бактериальных ансамблей ризосферы некоторых нетрадиционных для страны овощей, выращиваемых в условиях защищенного грунта на юге Западной Сибири.
Место и время исследования. Новосибирск, 2016.
Методология. В середине сентября в конце вегетационного сезона отобрали образцы почвы ризосферы растений бенинказы (Benincasa hispida), момордики (Momordica charantia), кивано (Cucumis metuliferus) и вигны (Vigna unguiculata), выращенных на торфяном почвосубстрате в условиях защищенного грунта теплицы, эксплуатируемой более 40 лет. Метагеномную ДНК экстрагировали, амплифицировали с помощью V3-V4 праймеров к гену 16S рРНК, и ампликоны секвенировали на Illumina MySeq. Полученные операционные таксономические единицы (ОТЕ) использовали для выявления функциональных групп с помощью базы данных FAPROTAX.
Основные результаты. В бактериобиоме ризосферы доминировали представители типов Proteobacteria (32±11% от общего числа последовательностей), Acidobacteria (23±7%) и Actinobacteria (18±3%), последовательности которых в сумме составляли три четверти обилия ризосферного бактериобиома. Всего было выявлено 20 типов бактерий. Биоразнообразие бактериобиома ризосферы оказалось очень высоко: так, индекс α-биоразнообразия Шеннона варьировал 7,0-7,5. Число выявленных ОТЕ (видовое разнообразие) было тоже высоко, варьируя по образцам 4500–4900, в то время как оценка потенциального числа ОТЕ (индекс Као-1) составила 5100–5700 ОТЕ на образец. Все ОТЕ были равномерно распределены в бактериобиоме, и индексы доминирования (Симпсона и Бергер-Паркера) были очень низки. Основной доминант, но всего с 1% относительного обилия, относился к семейству Bradyrhizobiaceae; всего было выявлено 27 ОТЕ этого семейства, однако FAPROTAX выявила всего четырех азотфиксаторов среди них. В целом выявленные функциональные группы указали на большой потенциал бактериальных ансамблей в плане процессов трансформации углерода, денитрификации, деградации ароматических соединений и растительных полимеров; патогенов растений не было выявлено. Наибольшая разница была выявлена между момордикой и остальными овощными культурами: в ризосфере момордики оказалось повышено относительное обилие Arthrobacter и Sphingomonas и число ОТЕ, способных к деградации ароматических соединений.
Заключение. Выявленное высокое биоразнообразие бактериобиома ризосферы овощных культур позволяет заключить, что эти овощные культуры успешно привлекали своими корневыми выделениями разнообразную микробиоту из тепличного торфяного почвосубстрата, что, в свою очередь, свидетельствует о высоком биоразнообразии микробиоты, существующей в почвосубстрате теплицы на протяжении ее длительной эксплуатации. Несмотря на сходство таксономической структуры бактериобиома ризосферы, выявленные особенности различных культур подтверждают необходимость включения изучения бактериального разнообразия в программы селекции и отработки технологий тепличного производства овощей.
Ключевые слова: микробиота ризосферы; секвенироваие ампликонов генов 16S рРНК; момордика; бенинказа; кивано; вигна; Западная Сибирь
Цитирование: Naumova N. B., Savenkov O.A., Alikina T.Yu., Fotev Yu.V. Characterization of the core bacteriobiome in the rhizosphere of greenhouse vegetables: taxonomic diversity and putative functions // Почвы и окружающая среда. 2020. Том 3. № 3. e128. doi:10.31251/pos.v3i3.128
Литература
Berendsen R.L., Pieterse C.M., Bakker P.A. The rhizosphere microbiome and plant health // Trends Plant Sci. 2012. V.17. No.8. P. 478–486.
Bier R.L., Bernhardt E.S., Boot C.M., Graham E.B., Hall E.K., Lennon J.T., Nemergut D.R., Osborne B.B., Ruiz-González C., Schimel J.P., Waldrop M.P., Wallenstein M. D. Linking microbial community structure and microbial processes: an empirical and conceptual overview // FEMS Microbiol. Ecol. 2015. V.91. No. 10. fiv113.
Edgar R.C. UPARSE: highly accurate OTU sequences from microbial amplicon reads // Nat Methods, 2013. V.10. P.996–998.
Edgar R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon reads // bioRxiv, 2016a.
Edgar R.C. SINTAX, a Simple Non-Bayesian Taxonomy Classifier for 16S and ITS Sequences // bioRxiv, 2016b.
Fadrosh D.W., Ma B., Gajer P., et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform // Microbiome. 2014. V.2. P.1–6.
Fang E.F., Froetscher L., Scheibye-Knudsen M., Bohr V.A., Wong J.H., Ng T.B. Emerging Antitumor Activities of the Bitter Melon (Momordica charantia) // Curr. Protein Peptide Sci. 2019. V.20. No. 3. P.296–301.
Floc'h J.B., Hamel C., Lupwayi N., Harker K.N., Hijri M., St-Arnaud M. Bacterial Communities of the Canola Rhizosphere: Network Analysis Reveals a Core Bacterium Shaping Microbial Interactions // Front Microbiol. 2020. V. 11. Article 1587.
Fotev Y.V., Syso A.I., Shevchuk O.M. Introduction in Siberia (Russia) of new vegetable species with a high biochemical value // PlantGen2019. Current. Challenges in Plant Genetics, Genomics, Bioinformatics, and Biotechnology. Proc. of the Fifth Inter. Sci. Conf. (Novosibirsk, 24-29 June, 2019). Kochetov A., Salina E. (eds.). Novosibirsk, 2019. 12–14.
de la Fuente Cantó C., Simonin M., King E., Moulin L., Bennett M.J., Castrillo G., Laplaze L. An extended root phenotype: the rhizosphere, its formation and impacts on plant fitness // The Plant J. 2020. V. 103. Iss. 3. P.951–964.
Hammer O., Harper D.A.T., Ryan P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis // Palaeontologia Electronica. 2001. V.4. Iss.1. P.9.
Huang H., Chen F., Long R., Huang G. The antioxidant activities in vivo of bitter gourd polysaccharide, // Int. J. Biol. Macromol. 2020. V.145. P.141–144.
Igolkina А.А., Grekhov G.A., Pershina E.V., et al. Identifying components of mixed and contaminated soil samples by detecting specific signatures of control 16S rRNA libraries // Ecol. Ind. 2018. V.94. P.446–453.
Kumar A., Dubei A. Rhizosphere microbiome: Engineering bacterial competitiveness for enhancing crop production // J. Adv. Res. 2020. V. 24. P.337–352.
Louca S., Parfrey L.W., Doebeli M. Decoupling function and taxonomy in the global ocean microbiome // Science. 2016. V. 353. P.1272–1277.
Marchesi J.R., Ravel J. The vocabulary of microbiome research: a proposal // Microbiome. 2015. V. 3. P.31.
Mazzola M. Assessment and management of soil microbial community structure for disease suppression // Annu Rev Phytopathol. 2004. V.42. P.35–59.
Pinton R., Varanini Z., Nannipieri P. The Rhizosphere: Biochemistry and Organic Substances at the Soil-Plant Interface: Boca Raton: CRC Press, 2007.
Rovira A.D. Plant root exudates // Botanical Review. 1959. V.35. No. 1. P.35–57.
Rovira A.D. Plant root excretions in relation to the rhizosphere effect. IV. Influence of plant species, age of plant, light, temperature, and calcium nutrition on exudation // Plant and Soil. 1969. V.11. P.53–64.
Singh R., Kumar A., Singh M., Vandana, Kumar Maurya S., Pandey K.D. Microbial Diversity in the Rhizosphere of Momordica charantia L. Bitter Gourd, Int. J. Curr. Microbiol. App. Sci. 2017. V.6. No. 2. P.67–76.
Sur S., Ray R.B. Bitter Melon (Momordica Charantia), a Nutraceutical Approach for Cancer Prevention and Therapy // Cancers. 2020. V.12. No. 8. Article 2064.
Tian Y., Gao L. Bacterial Diversity in the Rhizosphere of Cucumbers Grown in Soils Covering a Wide Range of Cucumber Cropping Histories and Environmental Conditions // Microb. Ecol. 2014. V.68. P.794–806.
Tian L., Lin X., Tian J., Ji L., Chen Y., Tran L. P., Tian C. Research Advances of Beneficial Microbiota Associated with Crop Plants // Int. J. Mol. Sci. 2020. V.21. No. 5. P.1792.
Trivedi P., Delgado-Baquerizo M., Trivedi C., Hamonts K., Anderson I.C., Singh B.K. Keystone microbial taxa regulate the invasion of a fungal pathogen in agro-ecosystems // Soil Biol. Biochem. 2017. V.111. P.10–14.
Trivedi P., Schenk P. M., Wallenstein M. D., Singh B. K. Tiny Microbes, Big Yields: enhancing food crop production with biological solutions // Microb. Biotechnol. 2017 V.10. No. 5. P.999–1003.
Uren N.C. Types, amounts, and possible functions of compounds released into the rhizosphere by soil-grown plants. In The Rhizosphere. CRC Press: Boca Raton, USA, 2000. P.35–56.
Vishwakarma K., Kumar N., Shandilya C., Mohapatra S., Bhayana S., Varma A. Revisiting Plant–Microbe Interactions and Microbial Consortia Application for Enhancing Sustainable Agriculture: A Review // Front. Microbiol. 2020. V. 11. Article 560406.
Wang Z., Zhang J., Wu F., Zhou X. Changes in rhizosphere microbial communities in potted cucumber seedlings treated with syringic acid // PLoS ONE. 2018. V. 13(6). e0200007.
Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naḯve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy // Appl. Environ. Microbiol. 2007. V.73. P.5261–5267.
Wen T., Yuan J., He X. et al. Enrichment of beneficial cucumber rhizosphere microbes mediated by organic acid secretion // Hortic. Res. 2020. V.7. P.154.
Yu D., Ma Y., Chen M., Dong X. KOH activation of wax gourd-derived carbon materials with high porosity and heteroatom content for aqueous or all-solid-state // J. Colloid Interface Sci. 2019. V.537. P.569–578.
Zhalnina K., Louie K.B., Hao Z., Mansoori N., da Rocha U.N., Shi S., Cho H., Karaoz U., Loqué D., Bowen B.P., Firestone M.K., Northen T.R., Brodie E.L. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly // Nat. Microbiol. 2018. V.3. No.4. P.470-480.
Zhao X., Jiang Y., Liu Q., Yang H., Wang Z., Zhang M. Effects of Drought-Tolerant Ea-DREB2B Transgenic Sugarcane on Bacterial Communities in Soil // Front Microbiol. 2020. V.11. Article 704.
Zhou Y., Ma Y., Zeng J., Duan L., Xue X., Wang H., Lin T., Liu Z., Zeng K., Zhong Y., Zhang S., Hu Q., Liu M., Zhang H., Reed J., Moses T., Liu X., Huang P., Qing Z., Liu X., … Huang, S. Convergence and divergence of bitterness biosynthesis and regulation in Cucurbitaceae, Nat. Plants. 2016. V.2. 16183.
Zhou J., Yu L., Zhang J., Zhang X., Xue Y., Liu J., Zou X. Characterization of the core microbiome in tobacco leaves during aging // Microbiology Open. 2020. V.9, No.3. e984.
Zhu M., Huang R., Wen P., Song Y., He B., Tan J., Hao H., Wang H. Structural characterization and immunological activity of pectin polysaccharide from kiwano (Cucumis metuliferus) peels // Carbohydrate polymers. 2021. V.254. Art. 117371.
Сведения о авторах:
Наумова Наталья Борисовна – кандидат биологических наук, ведущий научный сотрудник лаборатории агрохимии ФГБУН Институт почвоведения и агрохимии Сибирского отделения Российской академии наук (г. Новосибирск, Россия); naumova@issa-siberia.ru
Савенков Олег Александрович – ккандидат биологических наук, старший научный сотрудник лаборатории агрохимии ФГБУН Институт почвоведения и агрохимии Сибирского отделения Российской академии наук (г. Новосибирск, Россия); savenkov@issa-siberia.ru
Аликина Татьяна Юрьевна – младший научный сотрудник ЦКП «Геномика» ФГБУН Институт химической биологии и фундаментальной медицины Сибирского отделения Российской академии наук (г. Новосибирск, Россия); alikina@niboch.nsc.ru
Фотев Юрий Валентинович – кандидат сельскохозяйственных наук, старший научный сотрудник лаборатории интродукции пищевых растений ФГБУН Центральный Сибирский ботанический сад Сибирского отделения Российской академии наук (г. Новосибирск, Россия); fotev_2009@mail.ru
Авторы прочитали и одобрили окончательный вариант рукописи
Статья доступна по лицензии Creative Commons Attribution 4.0 License